|
|
.NET database and distributed computing tools |
|
|
|
||||
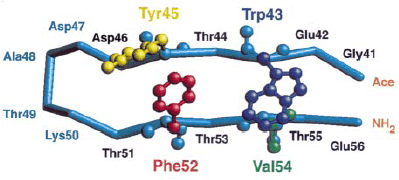 |
Fig. 1: Experimentally observed native shape of the ß-hairpin The three-dimensional structure of a single immunoglobulin binding domain (B1, which comprises 56 residues including the NH2-terminal Met) of protein G from group G Streptococcus, as determined in solution by nuclear magnetic resonance spectroscopy. |
| What makes the problem so difficult is the staggering amount of computation required to perform such simulations, which require calculating all the forces acting on all the atoms. Even for a relatively small protein, a single nanosecond of simulation may require a solid day of processing on a fast PC, and millions of such steps are required to observe the complete folding behavior of a single molecule with sufficient accuracy. | Simulating just one microsecond of folding can take months of supercomputing time. In fact, so much horsepower is needed that researchers have only recently been successful at simulating the folding of very short proteins. In 2001, Folding@Home set a record by using its volunteer network to simulate the first 38 microseconds in the folding of a snippet of protein (known as the beta-hairpin). The previous record was one microsecond, and that took several months on a Cray supercomputer. |
Fig. 2: Simulated folding of the ß-hairpin
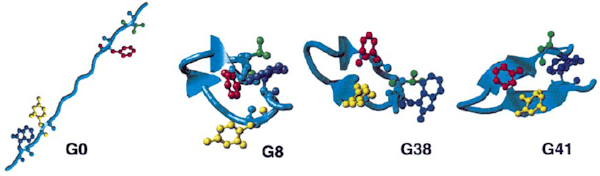
| A succession of snapshots from the folding trajectory for one member of the folded ensemble (out of 8 independent series, consisting of 100 Trial simulations each). At generation 41, one sees a cooperative formation of the fully formed hydrophobic core and of the key hydrogen bonds (Tyr45-Phe52, Trp43-Val54). | This event marks the completion of the folding process of the peptide and is accompanied by the RMSD, the total energy, and the total average number of hydrogen bonds all reaching their final, equilibrium values. After the peptide has folded, the hairpin structure remains fairly stable. |
| At first glance, it might appear that there is no good way to divide up this problem, since only one molecule is involved and interactions between all of its atoms must be taken into account. Furthermore, since each step inherently depends upon the previous step, there is no practical way to transform a sequential simulation into steps that can be performed entirely in parallel. Were it not for one very significant optimization, the collective power of all existing computers would not be enough to solve this problem by sheer, brute force.
A key insight in this particular case stems from the observation that the total time in such simulations is dominated by waiting for rare events that strongly influence what follows. |
Essentially, this means that most of the processing can be skipped for intermediate states between these significant "folding events",
vastly reducing the required amount of work and serial constraints of the problem.
The approach used by Folding @ Home is to give thousands of independent home PCs a molecular configuration with the same set of coordinates, but a different set of velocities for each atom. After some considerable time, when one PC finally succeeds at finding an acceptable lower-energy state, all of the other PCs are notified to stop working on this configuration, and begin another unit of work starting from the newly discovered configuration. An important feature of this algorithm is its near-linear scalability: the time it takes to predict the shape of a single protein is roughly proportional to the number of machines attacking the problem. |
Fig. 3: Stereoscopic comparison of simulation vs known native state of BBA5
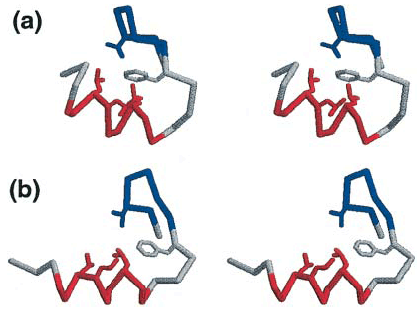
| Three-dimensional representation of a folded conformation (a) obtained by simulation, together with the experimentally determined native structure (b) of a small (23-amino acid) protein known as BBA5. With large-scale distributed computing, the multiplexed-replica exchange molecular dynamics (MREMD) method was tested starting from a fully unfolded state, | generating more than 200 microseconds of aggregate atomistic molecular dynamics simulation time. Overall, the conformation reached by simulation shows a good agreement with experiment, with well-defined secondary structure. (Relax your eyes to visualize these 3-D images.) |
| Of course, protein folding is just one particular example, and it isn't obvious how to use the same techniques in an entirely different scenario. Actually, though, similar optimizations can be applied to studying the dynamics of many kinds of complex systems and other types of "Monte Carlo" simulations. More generally, the
Virtual Supercomputing model lends itself well to optimizations where selective feedback from one process is used to refine the behavior of other processes, sometimes leading to dramatic speedups.
Storing intermediate values in a database for later consolidation is another good way to handle computationally intensive applications that seem to require a "giant main memory". In fact, the general technique of using a database to hold the results from multiple parallel processes has many commercial uses. |
This method, for example, can greatly accelerate long-running Monte Carlo simulations that analyze the exposure of a complex mix of investments, such as derivatives, to changing interest rates, inflation, exchange rates, portfolio diversity, etc.
Base One's grid computing software provides a solid foundation for building cost-effective solutions to very large scientific or business problems, without requiring dedicated, high-performance computers. Sometimes a little deep thinking is required, but there are all sorts of "tricks" that can turn an application into a promising candidate for grid computing. Once in place, the production system can be upgraded safely and continuously to handle additional supercomputing problems - each solved in its own "embarrassingly obvious" or occasionally, very clever way. |
References:
- Physicists Take on Challenge Of Showing How Proteins Fold, Bunk, S., The Scientist 12(21): 1 (10/26/1998)
- Home is where the fold is, The Economist Technology Quarterly (12/7/2000)
- Screen Savers of the World Unite!, Shirts, M. and Pande, V. S., Science Magazine, 290(5498): 1903-1904 (12/8/2000)
- Genome Effort Hits Home, Patrizio, A., Wired News (2/17/2001)
- Mathematical Analysis of Coupled Parallel Simulations, Shirts, M. R. and Pande, V. S., Phys. Rev. Letters, 86(22): 4983-4987 (5/28/2001)
- ß-Hairpin Folding Simulations in Atomistic Detail Using an Implicit Solvent Model, Zagrovic, B., Sorin, E. J. and Pande, V., J. Mol. Biol., 313: 151-169 (2001)
- Atomistic Protein Folding Simulations on the Submillisecond Time Scale Using Worldwide Distributed Computing, Pande, V. S., et al, Biopolymers, 68: 91-109 (published online in 2002)
- Folding@Home and Genome@Home: Using distributed computing to tackle previously intractable problems in computational biology, Larson, S. M., Snow, C. D., Shirts, M., and Pande, V. S., To appear in Computational Genomics, Richard Grant, editor, Horizon Press (2002)
- Simulation of Folding of a Small Alpha-helical Protein in Atomistic Detail using Worldwide-distributed Computing, Zagrovic, B., Snow, C. D., Shirts, M. R., and Pande, V. S., J. Mol. Biol. 323: 927-937 (2002)
- Native-like Mean Structure in the Unfolded Ensemble of Small Proteins, Zagrovic, B., Snow, C. D., Khaliq, S., Shirts, M. R., and Pande, V. S., J. Mol. Biol., 323: 153-164 (2002)
- Multiplexed-Replica Exchange Molecular Dynamics Method for Protein Folding Simulation, Rhee, Y. M. and Pande, V. S., Biophysical Journal, 84: 775-786 (2/2003)
- Insights Into Nucleic Acid Conformational Dynamics from Massively Parallel Stochastic Simulations, Sorin, E. J., Rhee, Y. M., Nakatani, B. J. & Pande, V. S., Biophysical Journal, 85: 790-803 (8/2003)
- Does Native State Topology Determine the RNA Folding Mechanism?, Sorin, E. J., Nakatani, B. J., Rhee, Y. M., Jayachandran, G., V Vishal, V., & Pande, V. S., J. Mol. Biol., 337: 789-797 (2004)
- Folding@Home: Can a grid of 100,000 CPUs tackle fundamental barriers in molecular simulation?, Pande, V., PARC Forum (video clip), (2/19/2004)
Selected links to the Folding@Home web site at Stanford University:
- Home page: http://www.stanford.edu/group/pandegroup/folding/
- Scientific background: http://www.stanford.edu/group/pandegroup/folding/science.html
- Recent research papers: http://www.stanford.edu/group/pandegroup/folding/papers.html
- The basics of Monte Carlo simulations: http://www.stanford.edu/group/pandegroup/folding/education/montecarlo/index.html
- Proteins: http://www.stanford.edu/group/pandegroup/folding/education/protein.html
- Simplified summary of Phys. Rev. Letters article of 5/28/2001: http://www.stanford.edu/group/pandegroup/folding/education/papers/phy_rev01.html
- Why not just run on a supercomputer?: http://www.stanford.edu/group/pandegroup/folding/faq.html#project.supercomputer
Distributed Computing | Benefits | Why Grid? | Architecture | In Depth | Queuing | Scalability | BFC
|
|
|||||||
| Home | Products | Consulting | Case Studies | Order | Contents | Contact | About Us |
|
|
|||||||
|
Copyright © 2012, Base One International Corporation |
|||||||